SELFIES, a new molecular representation method
I was thinking of writing my own explanation of SELFIES, partly because I couldn't find a detailed Japanese explanation of its grammar anywhere, and partly because I wanted to use it for my own research and wanted to deepen my understanding. Then, Mario Krenn, the first author, contacted me and said I could write about SELFIES on my blog. From there, I reread the paper a bit, examined the differences in behavior between the current version of SELFIES and this version, and came to an almost complete understanding of the grammar, which I wrote about SELFIES in Japanese in my blog.
And also I participated in the SELFIES Workshop on August 13, 2021.
accelerationconsortium.substack.com
Some of the participants asked me if there was an English version equivalent to the Japanese explanation I wrote. So I have translated it back into English.
Introduction
This article is an introduction to SELFIES, a new molecular representation presented in this paper, developed in the lab of Prof. It was developed in Prof. Aspuru-Guzik's lab, who is famous for creating Chemical VAEs. (It will be up on arXiv in 2019)
Original title: Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation
Authors: Mario Krenn, Florian Häse, AkshatKumar Nigam, Pascal Friederich, Alan Aspuru-Guzik
Journal: Machine Learning: Science and Technology
Publish: 28 October 2020
doi: 10.1088/2632-2153/aba947
I have written this paper for brevity and clarity, based not only on the paper, but also on my understanding of the behavior I have seen in running the code and the implementation, so please refer to the paper and the implementation if you are interested.
If you are interested, please refer to the papers and implementations. Also, the contents of this article are based on the latest version, v1.0.4.
[Note] Since the development has been going on since the paper was published, there are some discrepancies between the description in the paper and the output in v1.0.4. (I got stuck there...).
Outline of the paper
SMILES is widely used as a standard string molecular representation for chemical compounds, but when SMILES is used for input and output of generative models, there is a problem that a large part of the molecules do not correspond to valid molecules. The reason for this is that the generated strings are syntactically invalid or violate basic chemical rules such as the maximum number of valence bonds between atoms. The authors seem to have developed SELFIES to solve these problems.
Even a completely random string of SELFIES can represent a correct molecular graph. To achieve this, the authors developed SELFIES using ideas from theoretical computer science, namely formal grammar and formal automatons (formal Chomsky type-2 grammar (or analogously, a finite state automata)). In the paper, describe the development of SELFIES.
In the paper, we tried VAE and GAN with SELFIES and SMILES, and the results show that the output is fully valid and the model can generate orders of magnitude more molecules with SELFIES than with SMILES. (Table 1, Figure 5, Figure 6)
The great thing about SELFIES (in my opinion)
Instead of using strings to indicate the beginning and end of Rings and Branches, SELFIES represents Rings and Branches by their length, and avoids syntactic problems by interpreting the symbols following the Ring and Branch symbols as numbers representing their length (Index Symbols, to be discussed later).
Coding considering bond valence. It is designed to ensure that the physical constraint that C=C=C is possible (three carbons connected by a double bond), but F=O=F is impossible (F can only have one bond and O can only have two bonds) is met.
I have tried VAEs of Mol to SELFIES and Mol to SMILES using graph neural networks myself, and the generation of both SMILES and SELFIES works well, but most of the SMILES are not valid (several percent), while 100% of the SELFIES are valid. However, with SMILES, most of them are not valid (a few percent), but with SELFIES, they are 100% valid. (The details of the results of the test will be discussed another time.
Other results using SELFIES
Based on the results in the above paper, I believe that SELFIES will perform well in the task of inverse design of functional molecules based on deep generative models and genetic algorithms. We have already obtained a number of results using SELFIES.
- Inverse design using genetic algorithms
- Conversion from molecular images to SMILES
- Conversion from SMILES to IUPAC-name
The grammar of SELFIES (main topic)
About SMILES
If you don't know anything about SMILES, this article won't make any sense at all, but I'm going to assume that you do know something about SMILES.
If you don't know what SMILES is, I suggest you read py4chemoinformatics first to build up your domain knowledge, including how to use RDKit.
It's English version repository (original repository is here)
MDMA Explained (just look at it for now)
Now, let's start the explanation.
First of all, the SMILES of MDMA (3,4-Methylenedioxymethamphetamine), which appears in Figure 1 of the paper, is written as it is. MolFromSmiles("CNC(C)CC1=CC=CC=C2C(=C1)OCO2") in RDKit, it will be drawn as follows.

So, the SMILES of MDMA is CNC(C)CC1=CC=C2C(=C1)OCO2.
On the other hand, what happens to the SELFIES of MDMA?
import selfies as sf sf.encoder("CNC(C)CC1=CC=C2C(=C1)OCO2") # [C][N][C][Branch1_1][C][C][C][C][=C][C][=C][C][Branch1_2][Ring2][=C][Ring1][Branch1_2][O][C][O][Ring1][Branch1_2]
At this point, let's close the paper, because it's not the same as the expression of SELFIES in the paper (I know that the result is the same (it's just that this is how it was expressed in the version at the time)).
If you read Figure 2 properly, you may be able to understand it, but at first glance it doesn't make sense. First of all, the number of "C" seems to have increased compared to the original SMILES. It's hard to understand with MDMA, so I'll use a simpler example.
In order to understand the grammar of SELFIES, you need to understand Atomic Symbols, Index Symbols, Branch Symbols, and Ring Symbols. If you can understand these, you should have no problem.
Atomic Symbols
Let's start with an appropriate example.
sf.encoder("C=CC#C[13C]") # [C][=C][C][#C][13Cexpl] sf.encoder("CF") # [C][F] sf.encoder("COC=O") # [C][O][C][=O]
Atomic Symbols are composed of a bond type, which represents the bond type, and an atom type, which is represented by SMILES and denoted by .
The bond type is the same as in SMILES.
- single bond : '' (empty)
- double bond : '='
- Triple bond type: '#'
- Geometric isomerism: '/' and '\\'.
Atom type is the same as SMILES. However, if you explicitly use brackets (), such as [13C] or [C@@H], the character expl is added. Ions are also possible.
One of the reasons for the high Validity of SELFIES is the Decoder. Let's look at an example.
sf.encoder("COC=O") # [C][O][C][=O] sf.decoder("[C][O][C][=O]") # COC=O # Even if you ignore bond valence and write an appropriate compound, it will only be decoded to the part that is possible. sf.decoder("[C][O][=C][#O][C][F]") # COC=O
If the bond constraints of the previous or current atom are violated in this way, the bond multiplicity is designed to be reduced (minimally) so that all bond constraints are satisfied. (There is a description that gives the valence here: XXXXX_bond_constraints)
So far, I think you can understand the case of a simple linear chain. Next, I will explain the Branch Symbols and Ring Symbols, but before that, I would like to explain the Index Symbols, which are necessary for the explanation of Branch Symbols and Ring Symbols. This is because the Index Symbols, which are essential for the explanation of Branch Symbols and Ring Symbols, need to be explained first.
Index Symbols
This is where I personally got very stuck (and didn't notice it), but as I mentioned above, here are the key points.
- The beginning and end of a ring and branch in strings, SELFIES represents rings and branches by their length.
- After a ring and branch symbol, the subsequent symbol is interpreted as a number that stands for a length.
What this means is that you can think of index as a string that represents an atom, branching structure, or ring structure.
You can find the description here.
The table below shows the results.
| Index | Symbols |
|---|---|
| 0 | [C] |
| 1 | [Ring1] |
| 2 | [Ring2] |
| 3 | [Branch1_1] |
| 4 | [Branch1_2] |
| 5 | [Branch1_3] |
| 6 | [Branch2_1] |
| 7 | [Branch2_2] |
| 8 | [Branch2_3] |
| 9 | [O] |
| 10 | [N] |
| 11 | [=N] |
| 12 | [=C] |
| 13 | [#C] |
| 14 | [S] |
| 15 | [P] |
The symbol 𝑄 is used a lot in the paper, and this is what it means. Thinking in terms of hexadecimal numbers, 𝑄 can be expressed as 𝑄 = (idx(𝑠1)×162)+(idx(𝑠2)×16)+idx(𝑠3).
To see what I mean, let's take a look at the Branch Symbols example.
Branch Symbols
Now that we can use Index Symbols to calculate 𝑄, let's look at an example with a branch structure.
# SO4 sf.encoder("S(=O)(=O)([O-])[O-]") # [S][Branch1_2][C][=O][Branch1_2][C][=O][Branch1_1][C][O-expl][O-expl]
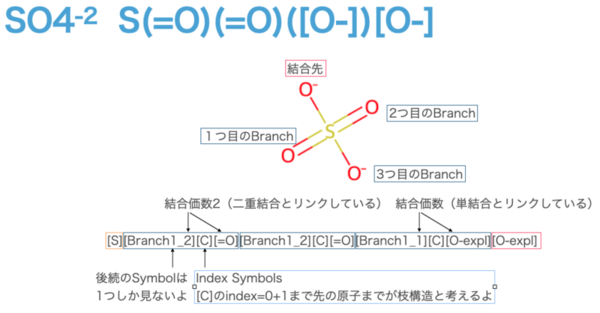
First, Branch Symbols has the pattern [Branch{L}_{M}]. The L on the left is the range {1, 2, 3}, and the number means how many subsequent letters are to be seen as Index Symbols. 1 means one letter, 2 means two letters, and so on. In this example, we are not looking at the second letter, so we need to assume that the second letter is a long chain, for example, "C" is more than 17 characters long (more on this later).
Next, the M on the right side of [Branch{L}_{M}] is the range {1, 2, 3}. This shows the bonding pattern with the atoms after the Index Symbols. 1 for a single bond, 2 for a double bond, 3 for a triple bond, and so on. Of course, the target Atom Symbols are linked to each other. By the way, if either one of them is not satisfied, the lesser one will be preferred, because the bond multiplicity is designed to be reduced (minimally) so that all bond constraints are satisfied if the bond constraints of the previous or current atom are violated.
If we consider the case of [Branch1_2] using SO4 as an example, only [C] will be processed as an Index Symbol, since subsequent Index Symbols will only see one character. In the table shown above, [C] has an index of 0. It is important to note that the branching structure is calculated from the Index Symbols, 𝑄, and is considered as a branching structure up to 𝑄+1 th symbol. In this case, all of them are [C], so the branch structure is up to the first letter after that ([=O] or [O-expl]). It is easy to understand that Atomic Symbols, without Branch Symbols, are directly connected to each other. (In this example, the head and tail are directly connected, and the other three are branch structures (orange and pink symbols).
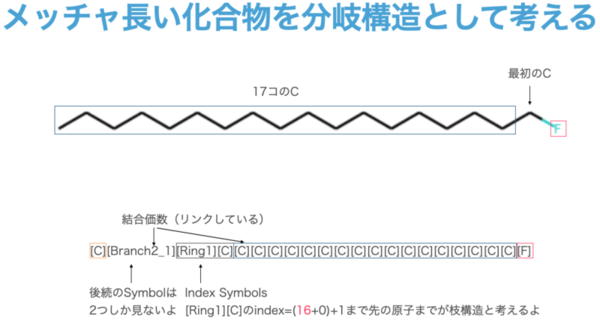
(In this example, the head and tail are directly connected, and the other three are branching structures. (Up to Branch 3 is possible, so something as long as 16 ** 3 is possible.
Ring Symbols
Now that you understand it, Ring Symbols is very easy.
There are two main patterns of Ring Symbols: [Ring] and [ExplRing], where L is {1, 2, 3} and the number of subsequent letters are considered as Index Symbols or not. Then, it calculates 𝑄 in the same way as Branch Symbols, and combines it with the 𝑄+1 th previous Atomic Symbols to form a ring structure.
Well, let's look at an example. Everyone's favorite :) Benzene rings are easy to understand.
sf.encoder("C1=CC=CC=C1") # [C][=C][C][=C][C][=C][Ring1][Branch1_2] # ↑ ↑ ↑ ↑ ↑ ↑ # 5 4 3 2 1 0
It is important to note that after [Ring1] are Index Symbols. You see [Branch1_2], but this is just what we think of as an index. Since it is [Ring1], only one character after it is considered as Index Symbols. Since [Branch1_2] has 𝑄 = 4 in the Index Symbols table, the first Atomic Symbols is [C], which is the 𝑄+1st, In other words, the five previous Atomic Symbol is the first [C], which is joined to form a ring structure.
Also, [ExplRing] about this pattern is easier to understand if you think of it as another representation of a benzene ring. The benzene ring can be written as C=1C=CC=CC=1 in SMILES. If we use this as SELFIES, we get the following.
sf.encoder("C=1C=CC=CC=1") # [C][C][=C][C][=C][C][Expl=Ring1][Branch1_2] # ↑ ↑ ↑ ↑ ↑ ↑ # 5 4 3 2 1 0
The basic idea is the same, but since [Expl=Ring1] is used, the bond type should be "=" when creating a ring structure with Atomic Symbols, which is the standard just before Ring Symbols. (You can imagine that =1 at the end of SMILES corresponds to [Expl=Ring1].
Extra for "Ring" use cases
Ring Symbols are a little more interesting, and triple bonds such as acetylene can be written with Ring Symbols. (In short, if you create a ring structure with a double bond in a part that already has a single bond, it will become a triple bond.
sf.decoder("[C][C][Expl=Ring1][C]") # C#C
MDMA Description
If you can understand this far, you will have a complete understanding of MDMA as shown earlier.
import selfies as sf sf.encoder("CNC(C)CC1=CC=C2C(=C1)OCO2") # [C][N][C][Branch1_1][C][C][C][C][=C][C][=C][C][Branch1_2][Ring2][=C][Ring1][Branch1_2][O][C][O][Ring1][Branch1_2]

It's a little difficult to understand because I couldn't color-code it well, but if you use all the assumptions you've made so far, you can understand it without any problems.
The difficult part is around the middle, [Branch1_2][Ring2][=C][Ring1][Branch1_2], where the branch structure is connected by a ring structure, which is quite difficult to understand, but if you think about it a bit, you can understand it.
Colab
This time, I used Colab because I wanted to run it in my hand so that I could do some thinking and error. It is available for viewing, so you can probably modify it in your own way or download it and try it out.
Summary
I hope that you now have an almost complete understanding of the syntax of SELFIES.
So why do SELFIES written in this format work so well for generative models such as VAEs and GANs, unlike SMILES? I think it is very important that SELFIES represents 100% valid compounds even if they are randomly sorted, that it is super robust, and that as a result, there are no invalid regions in the latent space.
I am wondering if I can use it successfully in my own research.
References
SELFIES GitHub github.com
SELFIES extensive blog post aspuru.substack.com
SELFIES Official Document selfies.readthedocs.io